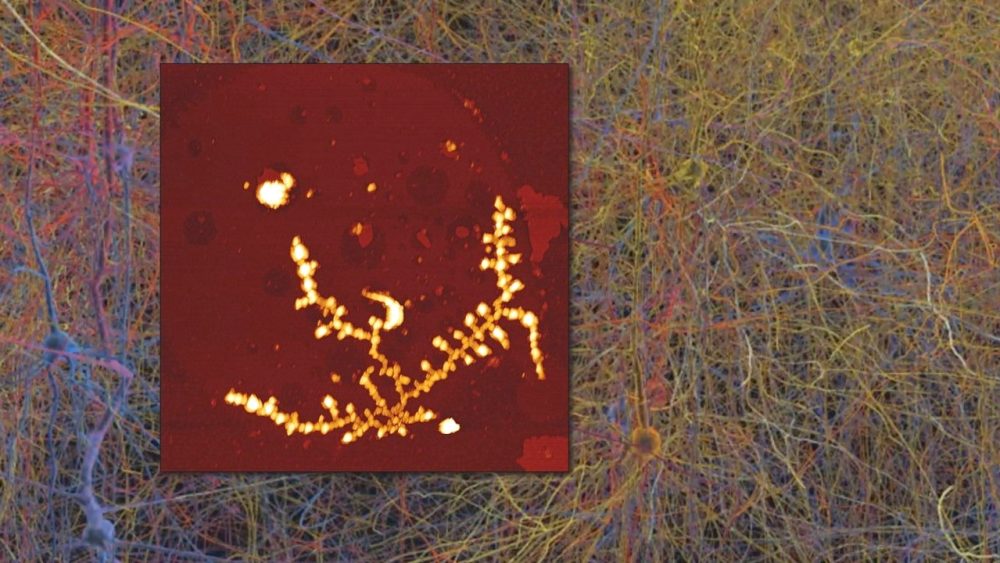
Protein-Verklumpung
ALS: Wenn Neuronen im Motorcortex durch fehlgefaltene Proteine sterben
Proteine spielen bei der Entstehung neurodegenerativer Krankheiten wie Parkinson oder der amyotrophen Lateralsklerose ALS eine wichtige Rolle. Sie entstehen im Zellinneren aus langen Ketten von Aminosäuren, die sich in spezifischer Weise dreidimensional falten. Kommt es in den sogenannten Motoneuronen, den im Hirn für die Bewegungssteuerung verantwortlichen Neuronen, bei bestimmten Proteinen zu Fehlfaltungen, dann kann das fatale Folgen haben: die Proteine beginnen zu verklumpen und führen schließlich zum Zelltod, der sich als Kettenreaktion bis in die Muskeln fortpflanzt. Die neuesten biomolekularen Forschungen sind den Ursachen dieser Protheinopathie auf der Spur und führen zu erfolgversprechenden Therapieansätzen dieser bisher kaum behandelbaren Muskellähmung.
Sprechertext der Sendung:
Manche bewegen sich extrem viel – andere sitzen eher ruhig am Schreibtisch herum. Aber selbst hier ist unsere Motorik gefragt. Wie stark der Mensch von seinem Bewegungssystem abhängt, fällt erst so richtig auf, wenn die Muskeln nicht mehr gut funktionieren. Die Symptome sind vielfältig und daher unter verschiedenen Krankheitsbezeichnungen bekannt: ALS oder Parkinson, ja sogar bei Alzheimer-Patienten kann es zu motorischen Defiziten kommen. Der Ursprung aller dieser neurodegenerativen Krankheiten liegt im Gehirn, und nach neuesten biomolekularen Forschungsergebnissen haben sie sogar vergleichbare Ursachen. Denn alle gehören inzwischen zur Klasse der „Proteinopathien“. Die Gewebeuntersuchungen von Erkrankten unter dem Mikroskop zeigen nämlich: In den Zellen kommt es zu typischen Veränderungen bestimmter Proteine.
Schrift-Einblendung:
Demenz: vor allem das Protein Tau und das Amyloid-beta Protein
Parkinson: vor allem das Protein alpha-Synuclein
ALS (amyotrophe Lateralsklerose): vor allem die Proteine SOD-1, TDP-43, FUS, C9orf72
Beispiel ALS: Alles Übel beginnt hier, in den besonders großen, hoch spezialisierten Zellen der sogenannten Motoneuronen. Dabei vor allem involviert: das ALS-Schlüsselprotein TDP-43.
O-Ton Christopher Secker, Mediziner, Charité Universitätsklinikum Berlin
Die Motoneuronen sind in der Großhirnrinde verortet und steuern von hier aus alle Muskelbewegungen im Körper. Zu dem sogenannten ersten Motoneuron gehört auch der weiter führende Nervenstrang, das sogenannte Axon. Es läuft durch das Rückenmark der Wirbelsäule. Hierdurch übertragen biochemische Botenstoffe, sogenannte Transmitter, die Nachrichten vom Gehirn weiter in den Körper. An unterschiedlichen Schnittpunkten erfolgt der Übergang vom ersten zum zweiten Motoneuron und von dort weiter zu den Muskelzellen. Was aber läuft falsch in den Motoneuronen, bevor sich die Krankheitssymptome von ALS zeigen? Über den genetischen Bauplan der DNA wird die Produktion der Eiweiß-Moleküle gesteuert. Zuerst entstehen lange Ketten von Aminosäuren. Sind die Elemente eines Proteins produziert, wird die lange Kette in eine komplizierte Struktur gefaltet. Jedes Protein hat eine dreidimensional genau festgelegte Oberflächenstruktur. Kommt es zu Fehlern in der Faltung solcher Proteine, kann das fatale Folgen haben. Bei den neurodegenerativen Erkrankungen diese: Die fehlgefalteten Proteine aneinander zu binden. Es entstehen große Protein-Aggregationen, die zunehmend die Funktionen in der Zelle stören. Zuletzt kollabiert die gesamte Zellfunktion, das betroffene Motoneuron stirbt ab.
Doch damit nicht genug. Die Proteinklumpen wandern auch aus der Zelle heraus.
O-Ton Christopher Secker, Mediziner, Charité Universitätsklinikum Berlin
Dieser Seeding-Prozess macht deutlich, dass TDP-43-Proteine nicht nur lokal in der Zelle aktiv sind. Sie binden sich an mRNA-Moleküle und wandern mit ihnen durch Axone, die Verbindungsstrecken von einer Synapse zur nächsten. Hier das Video eines in der Petrischale erzeugten Axons. Gelb eingefärbt ist das TDP-43 zu sehen. Die bei ALS bekannten, zur Fehlfaltung führenden Mutationen zeigen auch in den Axonen unterschiedliches Verhalten und neigen dazu zu aggregieren. Das behindert dann den mRNA-Transport durch die Axone und führt so zu einer Störung der Signalübertragung. Über diesen Transportweg können solche TDP-43-Klumpen übrigens bis zu gesunden Zellen im zentralen Nervensystem gelangen und dort dann auch pathogene Aggregationen des Proteins auslösen.
Dass solche Protein-Verklumpungen ein Merkmal unterschiedlicher neurodegenerativer Bewegungsstörungen darstellen, steht heute außer Frage. Bisher ist aber noch weitestgehend unbekannt, welche Aufgabe diesen Proteinen im gesunden Organismus zukommt. Vorläufig bleibt es daher die große wissenschaftliche Streitfrage, ob diese pathologisch veränderten Oberflächenstrukturen tatsächlich die Ursache der Erkrankung oder vielleicht nur Ergebnis eines anderen zellulären Auslösers sind. Das könnten beispielsweise fehlgesteuerte Reparaturmechanismen sein.
O-Ton Christopher Secker, Mediziner, Charité Universitätsklinikum Berlin
Solcher Ursachenforschung gehen Molekularbiologen und forschende Ärzte wie Christopher Secker in der Petrischale nach.
O-Ton Christopher Secker, Mediziner, Charité Universitätsklinikum Berlin
Am besten erforscht ist heute die erbliche ALS. Denn hier kennt man bereits Abschnitte in der DNA, die die Krankheit auslösen können. Allerdings ließ sich bisher nur bei etwa fünf Prozent von ALS-Erkrankten solch ein defekter Abschnitt im genetischen Bauplan feststellen. Die Forscher hoffen: Für erblich vorbelastete Menschen, die noch nicht an ALS erkrankt sind, könnte es daher bald eine prophylaktische Therapie geben.
O-Ton Prof. Dr. Thomas Meyer, Neurologe und Leiter der ALS-Ambulanz, Charité Universitätsklinikum Berlin
Die restlichen 95 Prozent der ALS-Erkrankten zeigen bisher jedoch keine dieser bekannten genetischen Veränderungen. Vielleicht liegt das aber nur an unserer Unkenntnis, denn neben dem jetzt entschlüsselten genetischen Bauplan gibt es noch viel im Genom zu erforschen.
O-Ton Prof. Dr. Thomas Meyer, Neurologe und Leiter der ALS-Ambulanz, Charité Universitätsklinikum Berlin
Aber was? Rund die Hälfte der 3,2 Milliarden DNA-Bausteine gehört zum sogenannt nicht-codierenden Teil unseres Erbgutes. Niemand weiß heute, welche biochemischen Effekte von diesem weitaus größeren, noch unentschlüsselten Bereich der DNA tatsächlich ausgehen. Lange galt dieser nicht-codierende Teil als bedeutungsloser DNA-Schrott. Diese Auffassung hat sich inzwischen deutlich geändert. Weltweite Forschungsanstrengungen laufen derzeit in einem großen zweiten Genomprojekt mit der Bezeichnung ENCODE. Nach den inzwischen erfolgreich entschlüsselten ersten drei Prozent des codierenden Bauplans sollen jetzt Schritt für Schritt auch die nicht-codierenden Teile des menschlichen Genoms entziffert werden. Das wird auch die Suche nach zellulären Krankheitsauslösern befruchten. Noch weiter in der Ferne liegt die Entschlüsselung des restlichen Anteils unseres Genoms, das die Genetiker auch „parasitäre DNA“ nennen, darunter 8 Prozent retroviralen Genmaterials. Diese HERV …
O-Ton Prof. Dr. Thomas Meyer, Neurologe und Leiter der ALS-Ambulanz, Charité Universitätsklinikum Berlin
Noch sind es nur sehr wenige hochspezialisierte Forschergruppen, die sich in die Wirkungsmechanismen dieser „Terra Incognita“ vorwagen. Der voraus blickende Wissenschaftler sieht aber grundsätzlich drei Möglichkeiten, wie solche Viren bei Erkrankungen mitspielen könnten.
O-Ton Prof. Dr. Thomas Meyer, Neurologe und Leiter der ALS-Ambulanz, Charité Universitätsklinikum Berlin
Trotz großer Fortschritte in der biomolekularen Forschung stehen wir also noch ganz am Anfang, die zellulären Ursachen neurodegenerativer Krankheiten zu verstehen. Doch trotzdem rechnet Thomas Meyer damit, dass bereits in den nächsten Jahren therapeutische Innovationen zu erwarten sind. Denn für eine wirksame Therapie muss man die biomolekularen Ursachen nicht unbedingt vollständig überblicken. Was der Medizinforscher für ALS erwartet, hat er mir in einem Telefoninterview so auf den strategischen Punkt gebracht:
O-Ton Prof. Dr. Thomas Meyer, Neurologe und Leiter der ALS-Ambulanz, Charité Universitätsklinikum Berlin
Erstsendung: November 2017
© 2017 mce mediacomeurope GmbH
Sämtliche Sendungen von HYPERRAUM.TV sind nur für die persönliche Information bestimmt und sind urheberrechtlich geschützt. Kopieren, Vervielfältigung, Bearbeitung, Verbreitung und jede Art der Verwertung außerhalb der Grenzen des Urheberrechtes sind nicht gestattet. Downloads und Kopien dieser Seite sind für den kommerziellen Gebrauch nicht gestattet.
